Next Generation Sequencing: A Gentle Introduction
posted on August 05, 2014DNA sequencing is the process by which we extract from raw cells a string of As, Cs, Ts, and Gs, known as a DNA sequence.
To understand why and how we are able to do this, it is helpful to understand a bit of the history of genetics.
A Brief and Incomplete History of the Gene
The idea of heredity has been around since at least 500BCE. Greek philosophers such as Anaxagoras, Aristotle, and Hippocrates all understood that, somehow, traits1 are passed from parents to their offspring. It wasn't until the mid-19th century when Gregor Mendel explicitly studied and formulated the basic principles of simple inheritance that the field of inherited variation was established.
Unfortunately, his work was largely disregarded and forgotten until the early 1900s when it was rediscovered and verified by several other scientists. Around the same time, Walther Flemming had developed a method of staining parts of cells, in particular the chromatin (which he named after this stain). With this technique he observed2 what would later be discovered to be dividing cells, or mitosis. Meanwhile, other scientists would discover the nucleobases of DNA3 (adenosine, thymine, guanine, cytosine), learn to isolate DNA4 (without knowing what it was), and guess at the structure of it using X-rays5.
The two bodies of work were unified in 1902 by Walter Sutton and Theodor Boveri in what is now known as the "Boveri–Sutton chromosome theory". Simply stated, chromosomes are replicated, inherited, and are the structural foundation of genetic inheritance.
In 1922, Hermann Muller published a paper6 speculating on the makeup of the gene. He states that they must be both autocatalytic and heterocatalytic—they direct both their own replication as well as the formation of other molecules. He also notes that viruses (barely known at the time) were essentially just packets of genes, and therefore that genes must be small (if many thousands of them were to fit on chromosomes), discrete packages of information that could be mutated & inherited. Not only that, but he also postulated that genes could be "side chains" of structured material which could interact in a mechanical way with the protoplasm to be replicated. In my mind7, Muller was the first to suppose that genes could be sequences of instructions used as a blueprint for creating life. However, many attribute this idea to Ewrin Schrödinger, who surely stated it with more certainty in his 1944 book What is Life?: "We believe the gene—or perhaps the whole chromosome fiber—to be an aperiodic solid."
It would be one of Muller's good friends along with one of his best students who in 1941 would formalize this idea into what is now one of the most important ideas of genetics, the "one gene-one enzyme hypothesis". Formulated by George Beadle and Edward Tatum, the hypothesis states that each gene corresponds with exactly one enzyme (in actuality, any type of protein, but they were close).
It bears brief mention that in 1953 James Watson and Francis Crick published a paper in Nature on the double-helical structure of DNA. More importantly, in my mind, was the "central dogma of molecular biology" that Crick stated in 1958: that information flows from DNA to RNA and then to proteins (but not from proteins to RNA or DNA).
The race was now on: how can we decode the information stored within DNA?
Early DNA Sequencing
Perhaps the first somewhat reasonable way of sequencing DNA was discovered by Ray Wu in 1970. It involved primers which would bind to specific portions of DNA; from this information, the actual sequence of the DNA could be, painstakingly, ascertained. A couple of viral phages were sequenced this way until a new and more efficient method of sequencing was devised in 1975 by Frederick Sanger8.
Sanger sequencing, as it is now known, was an ingenious breakthrough that, while not widely used today for sequencing human DNA, influenced and made practical the wide-scale study and use of DNA sequences. In essence, Sanger sequencing is four different experiments taking place at once. In each, all of A, T, C, and G, are added to the solution with DNA, and one special nucleobase (A*, C*, T*, or G*) which prevents the sequence from being extended any further once incorporated is also added. A bunch of DNA polymerase (a protein which constructs from a single strand of DNA the now famous paired double helix structure) is added to the solutions, and the reaction is allowed to run.
 the sequence](/images/autoradiogram.gif)
Because of these special terminating bases, the reaction randomly stops at a given point, leaving varying-length fragments of DNA all terminating with the same base behind.9 With enough DNA (and there's a lot: the reaction calls for many cells to be ground up and DNA shattered, the resulting fragments of DNA then extracted from the mix), and by marking all these special bases with a fluorescent particle, the positions of the given base are visible throughout the sequence. By running these fragments electrolytically through a gel we can order them by length, and read off the order of the bases. From this we get a sequence: a string of genetic information.
Sanger sequencing was used until about 2005, when it was supplanted by a new generation of sequencing technology which is collectively known as "Next Generation Sequencing" or "NGS".10
Sequencing: The Next Generation
Next Generation Sequencing isn't a single method, but rather a few different methods that all result in much higher throughput and lower prices for sequencing DNA. Prices are now as low as $1,00011 for a whole genome sequence, putting them well in the range of a clinical test. DNA sequencing is increasingly being used for, among other things, cancer screening (oncogenetics), as well as to determine reactions to and efficacy of certain drugs (pharmacogenetics).
Currently the most popular and widely used platform, Illumina12 sequences DNA by synthesis, using a combination of a modified shotgun sequencing approach and fluorescent dye on the nucleobases. Sequencing by synthesis is how Sanger sequencing worked: copying the single, denatured, strand of DNA with a polymerase. Shotgun sequencing13 is when we fragment the DNA into many small pieces prior to sequencing them (this was also done in Sanger sequencing). We call such fragments sequences "reads" or "short reads". They are spread all over a gel and amplified with PCR so that the florescent reaction which later takes place is more easily detected by the machine's camera.
Next, bases are added one at a time to the reaction, and if the next base in the strand being synthesized is the base added, it will emit a little *poof* of light which the machine's camera will pick up. Then the As (or Ts or Gs or Cs) are washed away, and another base is tried. If two or more of the same bases are added at once, the *poof* is bigger, and the camera records that too. The more bases in a row, though, the less accurate this gets.
This reaction is done in parallel, in many different strands on many different cells (little spots on the machine, basically, that DNA is attached to), so that it can be done significantly faster than it could be otherwise. The result of this sequencing is a library of short reads. This isn't very useful in itself; these reads must be assembled into a single sequence that is an individual's DNA.
It's important to note that many (probabilistically approaching all without enough coverage) reads will be overlapping with some other reads. This important property allows us to be reasonably sure where reads should map to a reference sequence.14 We use a reference sequence because it is much easier to map a given read to a reference (a map of the genome, really) with which it will match with over 99.9% of the time than it is to do what it called de novo assembly. De novo assembly is what we call aligning reads without any guidance at all. Much research is being done on faster and more accurate methods of de novo assembly, as it may be important for variant calling (I'll explain what that is later).
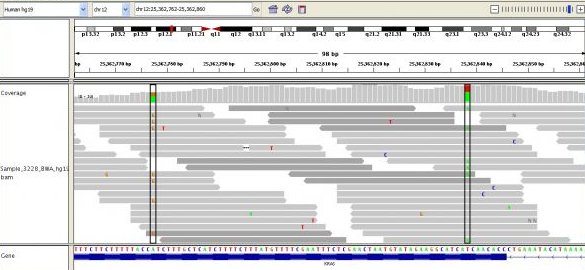
Above you can see many short reads (grey lines) overlapping one another and being mapped to the reference sequence, below. The deeper the average overlap of reads, the better the quality of the final sequence (in general). Read depth is also known as coverage, and you'll often see this referred to as, e.g., 10x or 40x or 100x. Some of the most sequenced genomes are up to 100,000x or more! The greater the coverage, though, the more expensive the sequencing, and the more time-consuming the alignment and subsequent analysis.
The output of these machines, FASTQ, is the first piece of data in a typical processing pipeline.
The NGS Pipeline: A Series of Tubes
Alignment: What Goes Where?
FASTQ files are very simple.15 They are essentially alternating lines of short reads (so yes, they really look like ACTATCGATAAAATGGACACGAGACGCGATTTTAAC and the like) and the estimated quality scores of those bases, in Phred format.16 Each line has a bit of metadata which tells which machine it was read from and a bit about where on the machine the sample was. I've created a simple example of a FASTQ file which you can peruse at your pleasure.
These FASTQ records are in no particular order. Alignment, or "realignment" as it is technically known when aligning against a reference sequence, is done with software such as BWA, iSAAC, or MAQ. Typically the reads are independently mapped to the reference sequence. The result of alignment is a SAM file (or the binary equivalent, BAM), short for Sequence Alignment/Map. This file contains many rows, each corresponding to a particular read. Each row contains the chromosome, position, and quality of the mapping for a given read, as well as a bit of information called the CIGAR string17 which explains how the read aligns to the reference. The reference string being mapped to may also appear in the row, or an MD tag18 may in its stead, which essentially carries the same information in a much smaller format, similar to the CIGAR string.
To get an idea of how big these files may get, a BAM file with 30x coverage of the entire genome could be as large as 175GB. Note that this is the compressed19 version of a SAM file. 30x coverage isn't that high, either.
So far I've not really mentioned how DNA can be mutated. This turns out to be very important for not only our phenotypes,20 but how we align and process genomics data. The three primary mutations we see are:
- Single nucleotide variants This is when the reference sequence says that there is an A, but the sample sequence shows a T, for example. A single nucleotide varies from the reference. A single nucleotide polymorphism, or SNP (pronounced "snip") is generally defined as a SNV which occurs in more than 1% of the population.
- Indels Or, insertion/deletions. This is when small segments (say, 50 or so base pairs [bps]) are either added or deleted from the sample sequence. For a small version, the reference sequence may say AATGTAGC, but our sample sequence may say AATGTAACCGGCCGC. These are a bit harder to detect, and can cause trouble when aligning sequences (but are nevertheless not uncommonly found in sequences).
- Structural variants (SVs) & copy number variants (CVs) There are either large indels (more than 50bps, and often degenerate repeats; one such CV causes Huntington's disease), or else completely wild splices such as when one chromosome splits and is reconnected to another. These aren't as uncommon as you might think, with some such mutations being a common cause of some cancers.
The CIGAR string encompasses the information describing the mutation's form. Aligners must account for these mutations so that they don't throw off the entire alignment process.
Mark Duplicates and Base Quality Score Recalibration: Making Sequences Suck Less
Now that we have an aligned BAM file, what can we do with it? Well, not much yet. Before anything else is done, we must process these BAM files a little bit further to improve their quality.
The first such step is called "marking duplicates" and serves to improve the quality of the data by removing reads which are exact duplicates of one another. Such reads can occur due to the PCR phase of sequencing preparation.21 They offer no additional information, they do not increase coverage; they are merely artifacts of the sequencing protocol. There is a non-zero probability that a duplicate removed is in fact not a true duplicate, but a read from a different part of the genome or even a fragment of DNA from another cell which shattered at the exact same spot. But, because the reads are at least one hundred base pairs long and because the genome is over 3.2 billion base pairs long, this is close to a statistical impossibility. Thus, we callously and perfunctorily remove such duplicates.
Next, we should perform what is known as base quality score recalibration (BQSR). In short, BQSR looks at all the bases in a sequence to determine a more accurate quality score for that base. A quality score, to be clear, is simply the probability that the base is actually what we're calling it. Low quality bases can occur due to the chemical properties of bases in certain configurations, bases near histones, the length of the read being sequenced (the further out we get, and the longer the chain, the lower quality the bases read), machine biases, and how near to the end of the chromosome the base is.22
Once these steps are done, we're ready to produce a useable product from all this incredibly difficult and time-consuming data-munging.
It's worth noting that these steps aren't actually necessary for processing, but generally can increase the outcome of the pipeline (depending on the assumptions the software run makes on the quality of the data and how it's been treated thus far). Some tools do many of these steps at once; some do just one step; some, none.23
Variant Calling: What's the Difference?
Variant calling is the art and science of determining the variants that are in the sample sequence. Or, really, determining what the sample sequence actually is. From this we can predict diseases, drug interactions, and conduct other research on the genome.
Variant callers essentially all look at pileup data,24 that is, the overlapping reads at a locus on the sequence, and determine if there is a variant there. If so, they attempt to determine what it is. Different variant callers can call different sorts of variants, with some specializing in SNVs, some in indels or SVs, or some which do all at once. Somatic variant callers are differentiated from normal (or "germline") variant callers in that they are focused on comparing two samples: normal tissue, and cancerous tissue (or other tissue of interest). Variant callers also often do what is sometimes a discrete step in the pipeline; indel (local) realignment. Indels can cause the alignment of reads to be off, and this can likewise cause variants to be called where they shouldn't be (and conversely). Thus, algorithms move the reads around (or change the CIGAR strings) based on what they see around a given locus.
The output of these callers is a file in variant call format, or VCF. This file format was created in 2008 by the 1000 Genome Project as a way to store variations between different genomes; important enough, it would seem, for their titular project.25
In fact, all file formats used in sequence processing and analysis have been rather more ad-hoc than VCF. SAM/BAM, as well as pileup format, were created by Heng Li and used in his samtools software library. FASTQ and FASTA (a simple textfile of a sequence) were settled on as industry standards which came about through convention. Another new project, ADAM, uses yet another format with the purpose of further compressing the data and simplifying the interoperation of different programs and tools. There will likely be many more formats in the future.
But I digress: back to VCFs.
VCF files contain information pertaining to the variants (or simply genotype; even if a locus doesn't vary from the reference, it may be included) in a given sample or samples. This includes an ID (which can either be a standard dbSNP ID, or an ID internal to the institution doing the processing), the position where the variant occurs, the type of variant, and other meta-information such as the genotype of the sample, the base quality, mapping quality, frequency of the allele in the pileups, and much, much more.
Now What?
What do we do with these VCFs? Well, it depends on who we are. If we're doctors, we may use it to guide our treatment (avoiding drug interactions, for example). We may use it to determine the type of cancer in the sample to guide treatment and adjust prognosis. Researchers are currently using them to create personalized cancer treatments, and to build vaccines that are specific to the specific cancer cells of an individual. Researchers can use them to determine the genes and mutations associated with diseases and other phenotypes, and thus guide research on cures and treatments for autoimmune, neurodegenerative, and other diseases which have long eluded cure.
Sequencing can also be applied to RNA, in order to determine gene expression in certain cells. We can also examine the epigenome, including methylation of DNA,26 which has been associated with aging27 and diseases such as cancer and HIV.
Some researchers are using sequencing to create and guide the creation of new, synthetic life. Others still use it for determining ancestry and the origin of our (and other) species.
There are many additional file formats such as GFF, ADAM, FASTA, and more. There are also many possible additional transformation which can be done to this sequencing data. The methods above may be the most common as of 2014, but new ideas are being tried every day.
There is a great wealth of information locked up in these data, and we are far from extracting all of it. In a very real way, our DNA sequence is the blueprint for the construction of our bodies. With a better understanding of our genome, microbiome, proteome, and interactome, environment, as well as the tools with which to act upon this knowledge, we will be able to cure and prevent diseases, enhance our species and others, and explore the makeup of life more adeptly than ever before.
-
"The structure of yeast nucleic acid P. A. Levene. ↩
-
Friedrich Miescher discovered it in the pus leftover from surgical gauze. ↩
-
"Variation Due to Change in the Individual Gene" H Muller. ↩
-
Many attribute this idea to Ewrin Schrödinger, who surely stated it with more certainty in his 1944 book What is Life?: "We believe the gene—or perhaps the whole chromosome fiber—to be an aperiodic solid". ↩
-
"A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase" F. Sanger, A.R. Coulson. ↩
-
This is an excellent illustrated explanation of the Sanger method. ↩
-
If we're being pedantic, shotgun sequencing has a slightly different meaning, as it has to do with a specific technology that is no longer in use, but the idea and effect are the same. ↩
-
A reference sequence is a sequence which is known to some high degree of certainty. They are created by resequencing the same DNA over and over, until the likelihood of an error at any given point is very low. There are few reference sequences used, and the are generally (with exceptions and caveats galore) trusted to be accurate. ↩
-
The CIGAR string is worth reading more about, and Matt Massie's blog is best place to go for it. ↩
-
The MD Tag in BAM Files is a great overview. ↩
-
Using, essentially, bzip. You may also generate indices for BAM files which can speed up access considerably. ↩
-
The phenotype is essentially the realization of our genome/epigenome/environment; e.g. our observable characteristics. ↩
-
This post goes over how duplicates arise in great details. ↩
-
Whereby a methyl group is added to the backbone of a nucleobase, c.f. wiki. ↩
-
Biomarkers and ageing: The clock-watcher. doi:10.1038/508168a ↩
-
There happens to be a next next generation of sequencing currently being researched, focusing on obtaining e.g. longer read lengths (which makes it easier to detect certain mutations called structural variants) and better error profiles, as well as more speed. Since, for the most part, these aren't being used yet, I'll just point you to the Wikipedia article explaining these new methods (they're very cool). ↩
-
Also known as phenotypes. ↩
-
It turns out that molecular biology is largely driven by tools. These tools are generally better ways of visualizaing the workings of cells and sub-cellular structures. Most great discoveries in biology are preceeded by great tools. ↩
-
Well, they are in theory as low as that. No genomics core that I'm aware of yet offers sequencing for near that price. ↩
-
Well, "must" is putting it a little strongly. There are various pipelines which implement various strategies for processing sequences. Not all require steps such as BQSR, but it's worth mentioned because, right now, it crops up a lot. ↩